C. acnes is the primary causative agent of acne vulgaris and other skin conditions. Acne vulgaris is a chronic inflammatory skin condition that affects about 9.4% of the global population, making it the eighth most prevalent skin disorder worldwide. C. acnes primarily resides within pilosebaceous follicles, adapting its metabolism to the host environment, and under anaerobic (hypoxic) conditions becomes more metabolically active and potentially virulent through complex host–microbe interactions. C. acnes is a predominant skin microbe that utilizes hydrolyzed triglycerides from sebum as an energy source. While many strains support the skin barrier through antioxidant activity and secretion of short-chain fatty acids, such as propionic acid, others can modulate the immune system and act as opportunistic pathogens.
CAcnesDB is a comprehensive up-to-date database of the C. acnes KPA171202 genome, hosting functional annotation from multiple levels of input and analyses, covering 2297 protein-coding genes. CAcnesDB stores hierarchical functional and structural characterization, i.e., modeled proteome, protein-protein interaction network, pocketome, and metabolome. To enable wide use of these data, CAcnesDB provides a suite of tools for searching, browsing, analyzing, and downloading the data. CAcnesDB also serves as a platform to add future genome annotations of related strains and perform a comprehensive analysis. By integrating a wide range of genomic data with tools for their use, CAcnesDB is a unique platform for both basic science research in C. acnes-related skin infections and disorders, as well as research into the discovery and development of drugs and possibly vaccines for acne vulgaris.
The genome of the C. acnes KPA171202 encodes 2297 protein-coding genes. Structural models were generated using ColabFold, based on the fast homology search of MMseqs2 for generating Multiple Sequence Alignments (MSAs) of the query protein, which is taken as input for the second algorithm, Alphafold2, to generate predicted structures, from which the best-ranked model is selected for each query protein. The scan ColabFold predicted models for 2280 proteins, nearly the whole proteome (excluding 17, of which 15 proteins have a polypeptide length greater than a thousand residues). The top-ranked predicted structures for each of the 2280 proteins constituted the C. acnes- structural proteome and were used for the prediction of the pocketome and associated ligands. The quality of the structural predictions from the ColabFold of the C. acnes proteome was analyzed. AlphaFold2, along with the actual three-dimensional coordinates, gives a measure of how confident one can be about the prediction itself, which is the predicted local distance difference test (pLDDT). Based on the reported AlphaFold thresholds, at the residue level, ColabFold generated high-confidence predictions for 67.4% of the C. acnes proteome, intermediate-confidence predictions for 18.9%, low-confidence predictions for 5.9%, and no prediction (i.e., pLDDT < 50) for 7.8%.
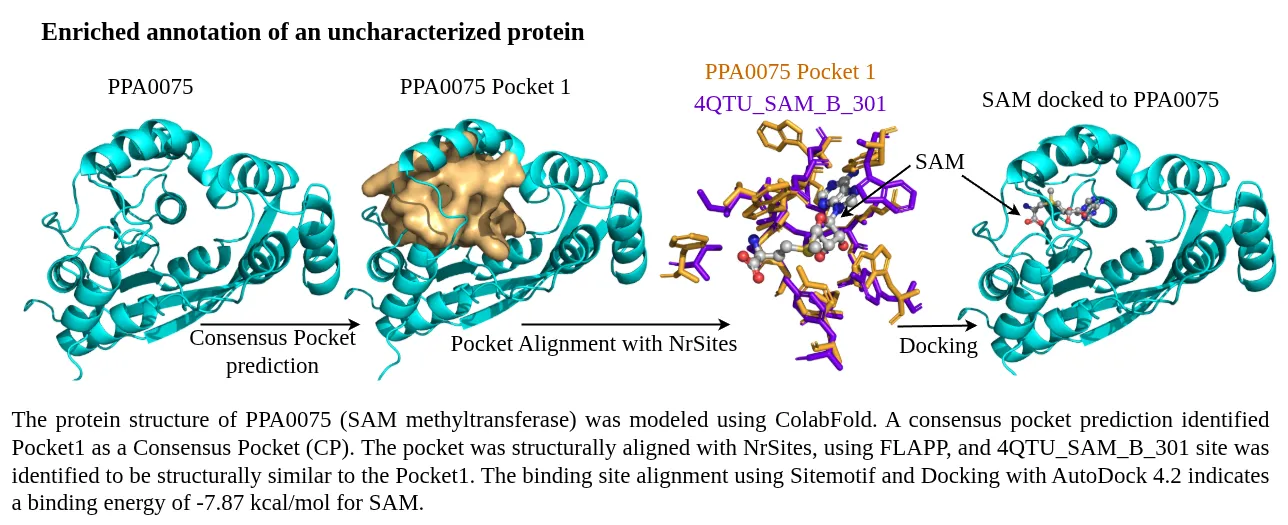
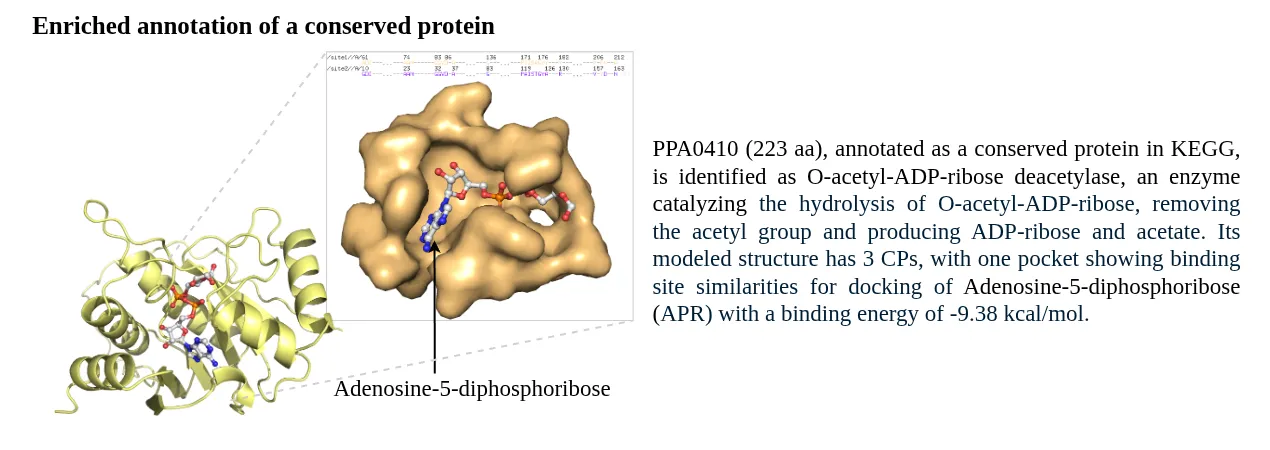
The top-ranked predicted structures for each of the 2280 proteins constituted the C. acnes- structural proteome and were used for the prediction of the pocketome and associated ligands. To predict possible small-molecule ligand binding pockets, three binding pocket prediction methods were used: Depth-based (i.e., PocketDepth), Energy-based (i.e., FPocket), and Voronoi tessellation-based (i.e., Site Hound). All three methods were run independently on each of the 2280 modelled structures (3 × 2280 = 6840). The numbers of pockets predicted by each of the methods are PocketDepth: 51,949, SiteHound: 22,800, and FPocket: 25,487. From these, consensus pockets (CP) were identified. The CPs from the FPocket output are used for this analysis, and only those FPocket pockets with at least 4 residues that match the pockets predicted by PocketDepth and SiteHound were considered further. Based on the pLDDT scores, the percentage of high and intermediate confidence residues (pLDDT ≥ 70) with respect to the total number of residues in the pocket was calculated to evaluate the quality of the individual pockets. Among all the AlphaFold pockets, about 77.58% are located in protein structure regions with relatively high and intermediate confidence (ratio > 0.7). This resulted in 11,266 CPs spanning over 2034 proteins, which together constitute the C. acnes’ Pocketome.

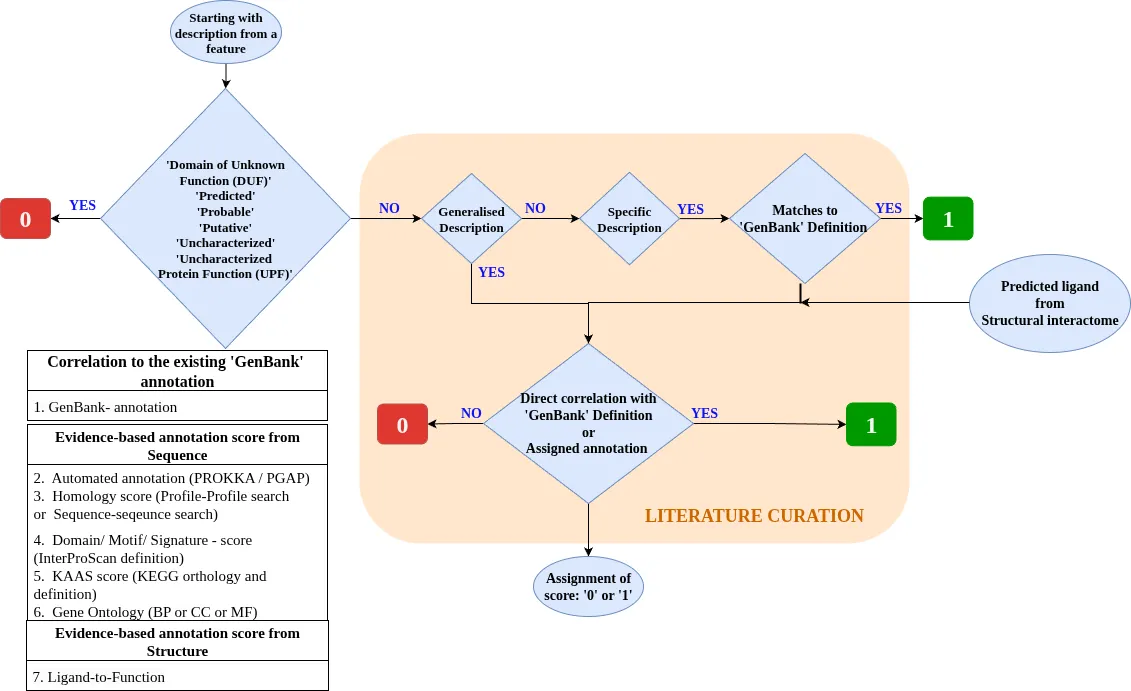
Iterative manual curation and literature-based up-to-date annotation were carried out to corroborate the assigned annotation across all the evidences for each individual protein. Additionally, an evidence scoring schema was devised to score the reliability of the assigned functional annotation. A binary-score schema was developed to evaluate the confidence of the assigned annotation. The flowchart for the computation of the annotation score is depicted in figure below.
-
The predicted function from each tool was manually checked, and a score of ‘1’ was given to the categories for which:
- Direct correlation was present from a particular piece of evidence.
- In case of multiple evidences, a Boolean logic of ‘either’ or ‘or’ was followed.
- If a particular description from an evidence suggests the possibility of correlation with a generalized term.
- Based on string match to the web search.
-
A score of ‘0’ was given to the categories for:
- The absence of any evidence.
- Any indirect correlation between the evidence and the assigned annotation.
- Based on the string match to ‘putative’, ‘uncharacterized’, or ‘Protein with the domain of unknown function (DUF)’.
- Cases of ‘indecision’ or ‘doubt’.
-
A special advantage was given to the literature-driven, experimentally characterized proteins.
A score of ‘5’ was assigned if the characterized protein has literature-based experimental validations.
For example, gene PPA1939 encodes the unique antioxidant-radical oxygenase of Propionibacterium acnes (RoxP), and the genes in the cluster PPA0859–PPA0865 are involved in Cutimycin biosynthesis.
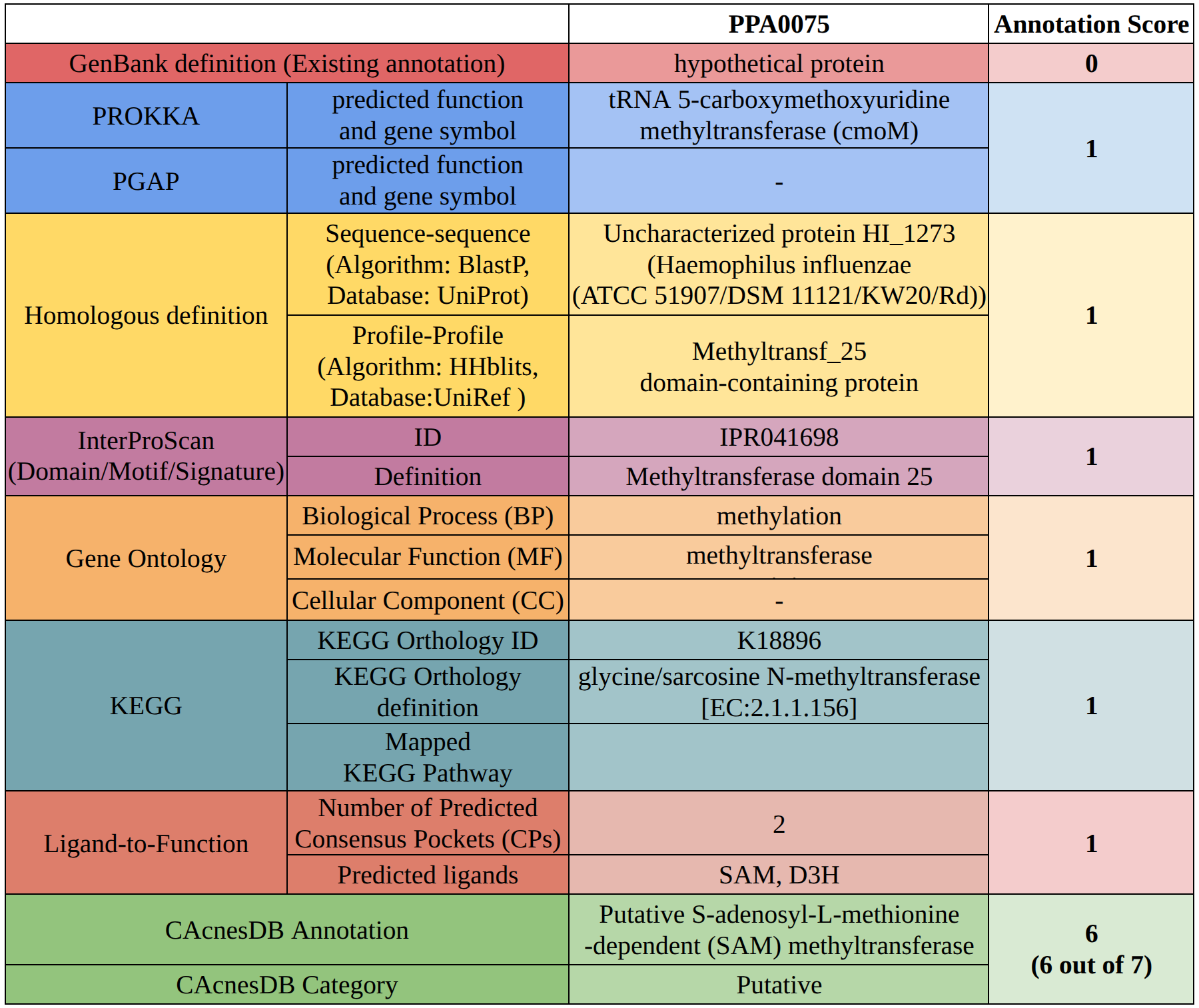
An example of the computation of the annotation score for the assigned annotation is depicted in the following figure.
STRING database was used to prepare a combined list of protein-protein interaction network, these included computationally predicted and experimental verified interactions. All the high-confidence interactions (combined score > 700) were retained.